1 Introduction
Traditional drug discovery generally involves some trial and error processes that include experimental screening of new chemical entities which are obtained by chemical synthesis or isolated from natural origin, until the desired pharmacological properties have been developed [1]. This traditional approach cost at least 1 billion US dollar and a period of at least 10 to 15 years to discover a successful drug [2]. Experts in the field of drug discovery and development have made several efforts to surmount the earlier mentioned challenges by coming up with the following methods: combinatorial chemistry methods, high throughput screening method, Proteomic and genomic projects, etc. Combinatorial chemistry has increased the number of compounds synthesized per given time, thereby populating the number of potential drug candidates to be screened. High throughput screening technique provided the opportunity to determine the biological potency of large chemical entities simultaneously. Advances in human genome and proteome have resulted in identification of large number of human proteins which serve as drug targets. All the contributions made by these techniques mainly caused an increase in expenses, number of leads and protein targets without corresponding increase in the number of successful new drug. Therefore, there is an urgent need for methods that will cut down cost and time for the drug discovery process. The use of computers and computer programs has emerged as an answer to this need in drug discovery, and is now known as computer aided drug design (CADD) [3].
In CADD, computational methods, mainly computer programs/algorithm, are employed to calculate structures and properties of molecules. These computational methods are broadly divided into two categories: molecular mechanics and quantum mechanics.
2 Computers in Medicinal Chemistry
2.1 Quantum Mechanics and Molecular Mechanics
Before molecular mechanics and quantum mechanics calculations and operations are carried out on molecules, chemical structure of the molecule in question has to be generated and displayed on the computer screen. Molecular modeling programs often include chemical drawing and graphics display packages capable of generating both two and three dimensional chemical structures. The chemical structures can be displayed in different formats as shown in Fig 1. There are several software packages available, such as ChemDraw, Alchemy, Sybyl, Hyperchem, Discovery Studio Pro, Spartan, CAChe, etc. [4].
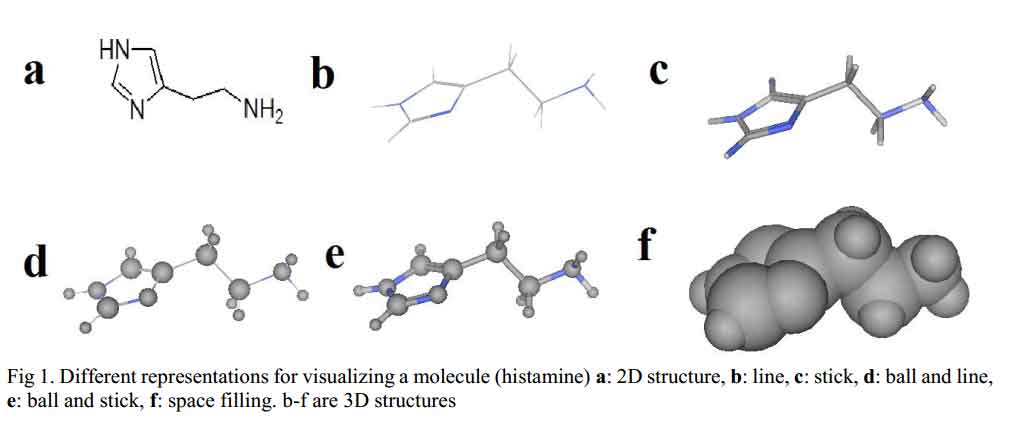
Click to view
2.2 Quantum Mechanics
Quantum mechanics uses the principles of quantum physics to calculate/describe the properties of a molecule by considering the interactions between the electrons and nuclei of the molecule. Electrons of a drug molecule are considered as the most important atomic particle in QM because the chemical behavior of a molecule (drug molecule phenomena) is governed by the probability of finding an electron in a particular location in the molecule and by the energy of that electron [5-6].
QM is based on molecular orbital theory (MOT) which enables the calculations/descriptions of electron location probabilities and energies following Schrodinger equation (1) on the potential energy surface.
![]()
Where H is the Hamiltonian operator for the system, E is energy (including the potential and kinetic energy components), ψ is the wave function which describes the distribution (3D coordinates) of an electron in a molecular orbital.
Solution to Schrodinger’s equation is only obtainable for the simplest molecule – hydrogen. Simplifications resulting in the following assumptions have to be made to the equation in order to make it tractable in the case of large molecules:
- Nuclei are regarded as motionless while electrons move around it. This assumption enables computation of electronic and nuclear energy to be made separately.
- The electrons move independent of each other, so the influence of other electrons and nuclei is taken as an average.
QM is used to calculate: molecular orbital energies and coefficient, heat of formation for specific conformations, partial atomic charges calculated from molecular orbital coefficients, electrostatic potentials. QM calculations are subdivided into three categories namely; ab initio, density functional theory and semi-empirical methods.
2.2.1 Ab Initio Methods
Ab initio is a Latin phrase meaning ‘from the beginning’. In this method, variation theory is used to compute energy of a molecule based on wave function. Full Schrodinger’s equation is used to treat all the electrons of a molecule without attempt to calibrate them against experimental data. Hartree-Fock model is an example of ab initio calculations.
Hartree and Fock combined electrons into an average field which simplified calculation by allowing Hamiltonian to be calculated for each electron independently using a new term for its interaction with the overall electron cloud.
Ab initio method implies the following:
- All the electrons have been considered simultaneously.
- The exact non-relativistic Hamiltonian (with fixed nuclei) is used.
![]()
Where the indices i, j and a, b refer, respectively, to the electrons and to the nuclei with nuclear charges Za and Zb. H is an “operator”, a mathematical construction that operates on the molecular orbital, ψ, to determine the energy.
- Lastly, an effort would have been made to evaluate all integrals rigorously.
Generally, ab initio method is satisfactory only for small molecules containing about tens of atoms. However, it has been applied to more computational work due to high computer horsepower. The method is used to determine properties of atoms of a molecule such as dipole moments, magnetic susceptibility, chemical shielding, spin-spin coupling constants, electron affinity, etc. GAMESS and GAMESS-UK are widely free academic software for carrying out ab initio calculations.
2.2.2 Density Functional Theory
Density Functional Theory (DFT) is QM method that calculates energy (electronic structure) of atoms of a molecule (especially the ground state) based on the electron density. Although DFT has been used to perform calculations in solid-state physics since the 1970s, it was only considered accurate enough for computations in quantum chemistry in the 1990s when the approximations in DFT were greatly refined to better model the exchange and correlation interactions. This placed DFT as a leading method used in describing electronic structure. However, the theory has not satisfactorily described intermolecular interactions like van dar Waals forces (dispersion), charge transfer excitations, global potential energy etc.
DFT method has been applied in different aspect of drug design processes such as: calculation of anion-binding properties of 2,6-diamidopyridin dipyrromethane hybrid macrocycles, analyzing the β2-adrenergic G protein-coupled receptor and predicting drug resistance of HIV-1 reverse transcriptase to nevirapine through mutations.
2.2.3 Semi Empirical Method
Semi empirical calculations were born out of necessity to solve the limitation of ab initio method i.e. computer intensity. Principally, semi empirical method is a simple Hartree Fock-Linear combination of atomic orbitals (HF-LCAO) based model that avoid all the difficult integrals, involved in ab initio method that makes it computationally intensive, which is usually calibrated against experiment. Semi empirical calculations are faster than ab initio method and uses perturbation theory to compute electronic properties such as electronic distribution and partial charges. This method is suitable for molecular systems containing hundreds of atoms. Molecular Orbital PACkage (MOPAC) and AM1 (Austin Model 1) are popular programs used in QM semi empirical calculations.
2.2.4 Quantum Mechanics in Drug Design (Electronic Charge)
Earlier, we have mentioned that MOT is the bedrock of QM methods. Molecular orbital calculations give numeric indices that show the electronic structure (probable position of an electron in a molecular orbital and its energy) which in turn governs the biological behavior/activity of a drug molecule. So, changes in numeric indices bring about change in electronic structure and invariably change in how a drug molecule behaves in vivo. Two examples that show the usefulness of MO calculations results (numerical indices) in interpretation of a drug’s mechanism of action and design of new drug molecule with improved properties are in the calculation of electronic charges.
The calculation of electronic charges reveals that electron charge density of atoms in a molecule is not evenly distributed. Valence electrons of atoms of a molecule are not localized on a particular atom rather they move around the entire molecule but spend more time nearer to electronegative atoms than electropositive ones. This results in some parts of the molecule being partially negative (due to excess electron) and other parts being partially positive (due to electron deficiency). Calculated electronic charges of molecules have been found useful in predicting structure-activity relationship of drugs as shown in the following.
Consider the three inhalation anesthetic gases shown in Fig 2 alongside their calculated excess or deficient electronic charges per atom of the molecule (not the absolute values). The electronic charge values enable drug scientist to suggest/propose the metabolism of these gases as thus: enzymatic ether bond cleavage by attack of an electrophilic oxygen atom at the methyl carbon in methoxyflurane is really much feasible than other two molecules because the methyl carbon is much less positive than the methyl carbons in others. The results of predictions like these are a better understanding of metabolism and a rationale for the design of new agents with improved properties.
Second example is gotten from the charge distribution of histamine and histamine ion (Fig 3) [7-8]. Charges are thought to be localized on a particular atom as the terminal nitrogen atom of histamine ion bears the positive charge. However, calculation of partial charges shows that some of the positive charge is localized on the hydrogen attached to the terminal nitrogen. This has important consequences in the way we think of ionic interactions between a drug and its binding site. It implies that charges areas in the binding site and the drug are more diffuse than one might think. This in turn, suggests that we have wider scope in designing novel drugs.
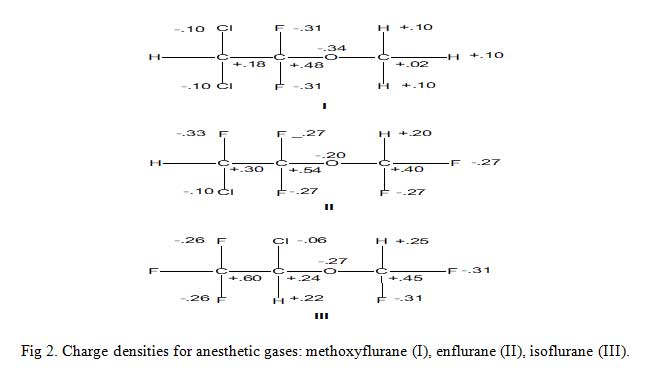
Click to view
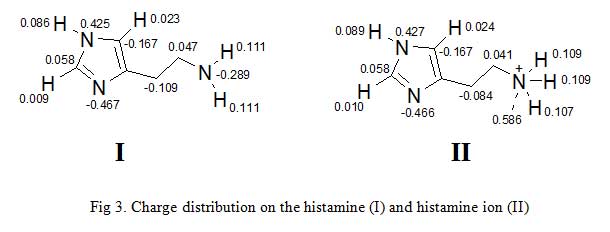
Click to view
2.3 Molecular Mechanics (MM)
In molecular mechanics, molecules are considered as a series of spheres (the atoms) connected by springs (the bonds). A large molecule consists of the same features we know about in small molecules, but combined in different ways. Equations, used in MM follow the laws of classical physics and are applied to nuclei without consideration of the electrons. The internal energies in MM are simply based on the Newtonian laws of classical mechanics. Equations derived from classical mechanics, are used to calculate the different interactions and energies (force fields) resulting from bond stretching, angle bending, non-bonded interactions, and torsional energies [9-10].
Etot = Estr + Ebend + Etor + Evdw + Eelec + … – – – – (3)
Where Etot is the total energy of the molecule, Estr is the bond-stretching energy term, Ebend is the angle-bending energy term, Etor is the torsional energy term, Evdw is the van dar Waal energy term, Eelec is the electrostatic energy term.
Values of bond length, bond angle, bond stretch and so on, at equilibrium are also known as force constants. These are used in the potential energy functions defined in the force field which describes a set known as force field parameters. The total energy of a molecule increases at any deviation from these equilibrium values. Each conformation of a molecule has its own total energy and a difference between the total energy of two different conformations of the same molecule is used to determine molecular stability.
2.3.1 Bond-stretching
If we consider histamine (Fig 4) we can identify a variety of bond types including C(sp2)-C(sp2), C(sp2)-C(sp3), and so on [11].
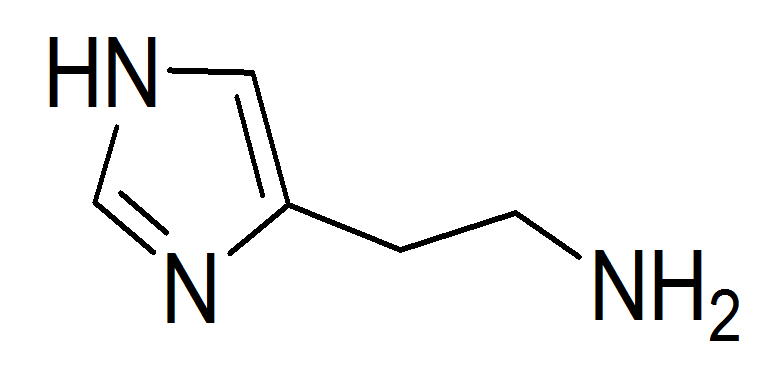
Click to view
There is usually an energy (interatomic force) change when the bonds stretch and contract from their ideal unstrained length. This bond-stretch energy term is described by the equation given below.
![]()
Where Kb is the bond-stretching force constant, ![]()
![]() bo is the unstrained bond length, and b
bo is the unstrained bond length, and b![]()
![]() is the actual bond length.
is the actual bond length.
2.3.2 Bond-bending/angle
Next, we have to consider the angle-bending vibrations. It is usual to write these as harmonic ones, typically for the connected atoms A-B-C.
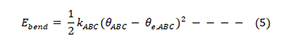
KABC (K is the angle-bending force constant, θe,ABC is the equilibrium value for the bond angle θ, and θABC is the actual value for θ.
2.3.3 Torsion angle
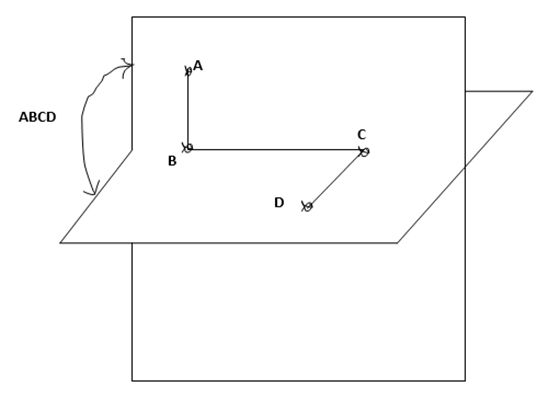
Torsional energies are associated with atoms that are separated from each other by three bonds. The relative orientation of these atoms is defined by the dihedral or torsion angle. The torsional angle ABCD between the four bonded atoms A, B, C and D is shown in Fig 5 [10].
Click to view
If we use χ to denote the angle between the four atoms, then a popular dihedral potential energy term is a cosine series given by
![]()
Where Kj is the torsional barrier, X is the actual torsional angle, the periodicity parameter, which would be 3 for a methyl group. Xe is the equilibrium (reference) torsional angle.
2.3.4 Non bonded interactions
MM force fields have to be transferable from molecule to molecule, therefore, the necessity of non-bonded interactions. These are usually subdivided into two types; Lennard-Jones and Coulomb’s interactions [5, 12-13].
2.3.5 Lennard-Jones interaction
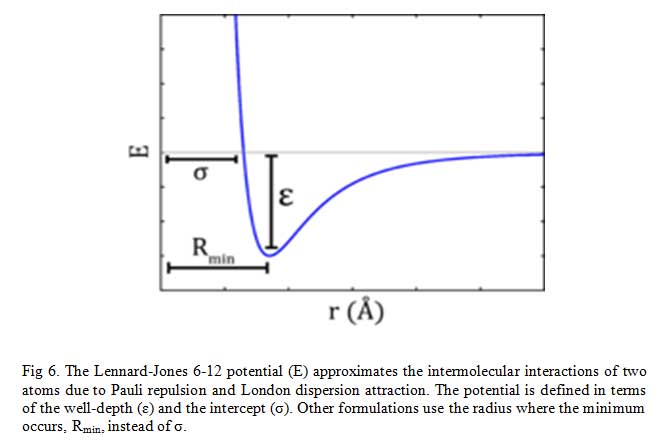
The Lennard-Jones potential as shown in Fig 6, describes the interactions of two neutral particles using a relatively simple mathematical model. Two neutral molecules feel both attractive and repulsive forces based on their relative proximity and polarizability. The sum of these forces gives rise to the van dar Waal interactions usually represented as Lenard-Jones potential (V(R) or E), as seen below:
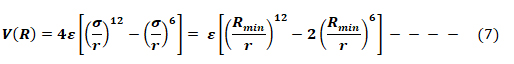
Where ε is the potential well depth, σ is the distance where the potential equals zero (also double the van-der-Waals radius of the atom), and Rmin is the distance where the potential reaches a minimum, i.e. the equilibrium position of the two particles.
The resulting curve from this equation looks very similar to the potential energy curve of a bond.
Click to view
2.3.6 Coulomb’s interaction
Coulomb interactions or electrostatic forces are involved in attraction or repulsion of particles or objects because of their net electric charge [5]. Coulomb noticed that forces acted along the line joining the centers of two charged bodies Qa and Qb, and that the forces were either attractive or repulsive depending on whether the charges were different or of the same type. The sign of the product of the charges therefore determines the direction of the forces.
![]()
Where Qa and Qb are the atomic charges of the interacting atoms, r is the distance between the charged bodies and K is a proportionality constant taken to be 1/(4πϵ0), where the permittivity of free space ϵ0 is an experimental determined quantity with the approximate value ϵ0 -8.854 x 10-12 C2N-1m-2.
The non-bonded term comprising Eelec and Evdw are a function of the distance between atom pair rij (non-bond cutoff distance).
To summarize the concept of MM, it is worthy to note the following;
- The energies calculated by MM are of no meaning as absolute quantity. They are only of relevance when compared to the energies of another conformation of the same molecule.
- MM calculations make use of data or parameters stored in tables within the program and that describe interactions between different sets of atoms.
- MM is used to calculate: energies of a molecule’s conformation, energy minimization and energies of a molecular trajectory/motion.
- MM is fast and less intensive on computer time relative to quantum mechanics.
- Lastly, because MM does not consider electrons, it cannot calculate electronic properties.
2.3.7 Example of MM in Drug Design (Energy Minimization)
Molecular mechanics calculations are applied in several aspects of drug design including energy minimization, docking, molecular dynamics, etc. Energy minimization will only be considered.
Energy minimization (EM) is a process by which stable, low-energy conformations of a molecule are calculated using the MM program/approach. EM is often performed to avoid atomic clashes and locate most stable conformation of molecules (Fig 7) [5, 14]. There is a possibility of unfavorable bonded and non-bonded interactions existing in a newly generated chemical structure. MM program calculates the energy of the new molecule then varies the bond lengths, bond angles, and torsion angles (this changes the geometry of the structure) and calculates the energy of the later structure. Comparison of the two energies of the first and later structure will show if a slight alteration in bond length or bond angle has effect on the overall energy of the molecule. The MM program will perform more geometrical changes and eventually stop at a structure in which geometrical variation result in only slight changes in energy – an energy minimum.
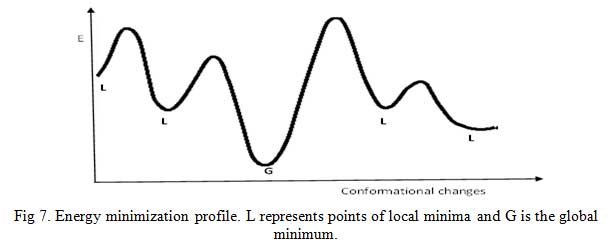
Click to view
2.4 Structure-Based Drug Design
Structure-based drug design (SBDD), also known as receptor-based drug design, is used when receptor (mainly enzymes/proteins) and ligand (small-molecule or drug) structures are both known [15]. The structure of the receptor can be determined by experimental methods such as X-ray crystallography or NMR. Alternatively, computational techniques such as threading or homology modeling can also be applied in obtaining structure of proteins whose structures are unavailable [16]. Then the binding site of the receptor is determined by either experimentally or computationally. In most cases, the protein is co-crystallized with a ligand. So the pocket where the co-crystallized ligand is located is considered the binding site of the protein [17]. A well characterized protein binding site, such as the interaction between the amino acids at the binding site of the protein and the ligand, can give information vital in designing of novel ligands or docking of putative ligand molecules.
The selective binding which occurs between small molecule ligand and a specific protein target is the basis of physiological activity and pharmacological actions of the ligand. Structural and energetic factors govern this binding event and are respectively captured in computational techniques by the prediction of binding mode/conformation (docking) and scoring of protein-ligand complexes.
2.4.1 Predicting Binding Mode – Docking
Docking is a computational technique used to predict, with a substantial degree of accuracy, the conformation (binding mode/pose) of ligands within the appropriate target-protein binding site [18]. It constitutes, therefore, a major technique employed in virtual screening by structure-based drug design [19]. Docking searches for different energetically permitted binding poses of a ligand at the protein active site by performing a number of trials. At the end of the trial searches, a pose is retained based on the calculated receptor-ligand interaction energy (score) of that pose. Conventionally, several poses of a molecule is generated by docking method and the score of each pose calculated using a scoring function usually affinity scoring function (Fig 8). The dock-score of each pose of the molecule is only of importance to medicinal chemist in comparison to other, that is, when prioritized. Placing the score in decreasing order is termed ranking. The pose with the lowest theoretical binding affinity is considered the best [20].
2.4.2 Scoring and Scoring Function
Almost all available scoring functions can be grouped into two categories: knowledge-based scoring functions and energy component methods [21]. In knowledge-based scoring function, statistical tools are used to compute the interatomic contact frequencies and/or distances in a database of crystal structures of protein-ligand complexes. Ligand binding affinity towards the target protein is assumed to be favored by molecular interactions that are close to the frequency maxima of the interactions in the database and vice versa. The observed frequency distributions are converted to what is called mean force or knowledge-based potentials. PMF, DrugScore, SmoG and Bleep are examples of knowledge-based potentials that predict binding affinity [22-24].
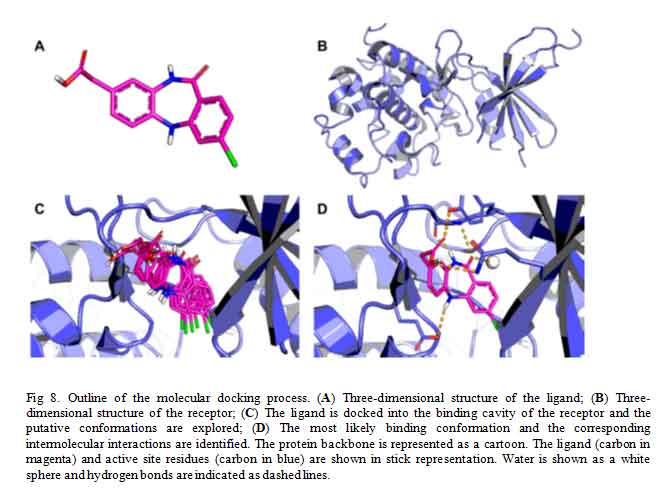
Click to view
They mainly differ in the type of molecular interaction that were considered and in the size of the training database used.
Scoring functions based on the energy component methods assume that the change in free energy upon binding of a ligand to its target can be decomposed into a sum of individual contributions:
ΔGbind = ΔGint + ΔGsolv + ΔGconf + ΔGmotion – – – – – – – – – -(9)
Where ΔGint is the specific ligand-receptor interactions, ΔGsolv is the interactions of ligand and receptor with solvent, ΔGconf is the conformational changes in the ligand and receptor and ΔGmotion is the protein and the ligand motion during the complex formation
Several applications using these methods to predict binding affinity have been developed such as LUDI, ChemScore, Validate, GOLD score, PLP, FlexX score, ScreenScore, AutoDock3 and so on [24].
2.4.3 Effect of Water-Solvation Energy
The biological system is made up of 70 % of water molecules. These water molecules play essential roles in the formation of protein-ligand complex in number of ways: They can mediate the contact between protein and a small molecule ligand by providing additional hydrogen bonds to the ligand. They can promote adaptability by allowing for promiscuous/ off-target ligand binding due to steric constraint. Consequently, the displacement of these water molecules by appropriate ligand functional moieties may be favourable to protein-ligand complex formation. Therefore, docking method which recognizes the explicit effects of structural water molecules and water-mediated interactions is highly desirable. Examples of such docking software are FlexX and SLIDE, etc [25].
Another method of drug design based on the knowledge of a biological target (structure-based drug design) is de novo method [26]. De novo design techniques are used when receptor structure is known and ligand structures are unknown. In this method, novel pharmaceutical active agents capable of interacting with a given receptor are computationally generated based entirely on the knowledge of the protein binding site. The proposed de novo model can be used to search large databases to identify compound fragments that can interact with specific sites in the receptor. GROW and LEGEND are examples of programs used in these techniques.
2.5 Molecular Dynamics
Target proteins are generally kept static while ligands are allowed to move about in docking. This is known as fixed-target-flexible-ligand docking and it is the type characterized in almost all virtual screening [27]. However, we know that biomolecules are dynamic in nature; therefore, docking is insufficient tool in computational predictions [28]. Structural dynamism of targets is accounted for through generations of multiple conformations of target by molecular dynamic techniques, Monte Carlo sampling, simulated annealing and even NMR [29].
In reality, atoms of molecules are never stable. Therefore, it is paramount to account for structural dynamism in attempt to describe biochemical systems. Dynamism of atoms of molecules in biological processes is recognized/computated by performing conformational sampling [30]. In molecular dynamic calculation, Newton second-order equation of motion (10) is solved for atom i with mass mi typically within a system of interacting forces and subjected to a net force Fi.
![]()
![]() is the second derivative of the positional vector calculated with respect to time. MD simulation is a deterministic method. Exact solutions for state properties can be derived from MD simulation at a given time and after specifying the initial set of system conditions. Once the starting atomic positions have been specified (typically obtained from x-ray crystallography and NMR spectroscopy), velocities are assigned according to a Maxwellian distribution as given in equation 11.
is the second derivative of the positional vector calculated with respect to time. MD simulation is a deterministic method. Exact solutions for state properties can be derived from MD simulation at a given time and after specifying the initial set of system conditions. Once the starting atomic positions have been specified (typically obtained from x-ray crystallography and NMR spectroscopy), velocities are assigned according to a Maxwellian distribution as given in equation 11.
![]()
Where P(v) is the probability, m and v are respectively the atomic mass and velocity, while k is the Boltzmann constant. According to the equipartition theorem, the system temperature T is related to the velocities (12) and (13),
![]()
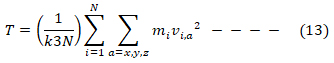
with the system kinetic energy, represented by Ekin and a representing the xyz coordinates. In principle, by correctly assigning the temperature T, according to the Maxwellian distribution the system under study becomes capable of dynamically evolving in a fashion similar to real life systems undergoing thermal motion [31].
Nowadays, one of the many applications of MD simulation technique in drug design and development is in investigating both structural and temporal stability of drug-receptor complexes under modeled experimental conditions such as solvent system, ionic concentration, temperature, and pressure. This becomes increasingly important in investigating stability of ligand-receptor complex as predicted by docking because the mere occurrence of binding may not always indicate the survival of such interaction on a time scale that is sufficient for altering physiological responses [32]. The predicted drug-receptor complex from docking calculation is considered stable if MD-generated drug conformations do not deviate by more than a given root mean standard deviation (rmsd), usually 2-3 Å. Additionally, MD has been applied to sample potential conformational states for a molecular target that has no suitable available crystallographic structures (structures with inaccessible or poorly defined binding sites). These samples conformations of the target (with accessible and well defined binding site cavities) can then be selected for molecular docking. Lastly, when a drug binds to a receptor, complex structure (drug-receptor complex) is formed which is more stable and causes equilibrium to shift towards the minimum energy complex structure. MD technique can be used to alternatively produce conformational states corresponding to these ligand-induced structures.
2.6 Ligand-Based Drug Design
When a receptor for a disease is unknown or the 3D structure is unavailable but a single active molecule is known then similarity searching is carried out in what is known as ligand-based virtual screening. In a situation where several actives are available then it may be possible to identify a common 3D pharmacophore, followed by a 3D database search [33]. If a reasonable number of active and inactive structures are known they can be used to train a machine learning technique such as a neural network which can then be used for virtual screening.
2.7 Pharmacophoric Screening
Ehrlich first defined pharmacophore as “a molecular framework that carries the essential features responsible for a drug’s biological activity” [34]. In pharmacophore-based screening, a typical of ligand-based drug design, a pharmacophore model is built which consist of how the positions of key amino acids will be in the active site of a target protein, feature type and direction of an active ligand. For instance, a key amino acid that acts as hydrogen bond donor should be in the location of a hydrogen bond acceptor feature in the pharmacophore model. A pharmacophore modeling cuts across ligand-based and structure-based. If the model is developed based on the knowledge of the ligand i.e. where several different known active molecules are used to identify the common important features, or from the target protein structure. However, pharmacophore model is categorized as structure-based when it is built based on the knowledge of the protein target structure [35].
2.8 SAR, QSAR and 3D-QSAR
Structure-Activity Relationship (SAR) approach attempts to explain the biological activity of a drug molecule as dependent on its molecular structure. Whereas Quantitative Structure-Activity Relationship (QSAR) moves further to examine the actual structure, characterize and quantify their physicochemical properties in numeric indices as parallel to their biological activity [36]. Crum-Brown and Fraser [37] were the first to publish equation in the field of QSAR, which set forth the idea that the biological activity of a compound Φ is a function of its structural properties C.
![]()
To account for the effect of 3D molecular shape to biological activity, 3D-QSAR was developed and added to QSAR models [38]. All of these efforts by SAR, QSAR and 3D-QSAR enable scientists involved in drug research to suggest mechanism of drug action and make predictions of more profitable areas for drug synthesis. This means that QSAR models allow the calculation of biological properties of novel analogues in advance so that only the ones with improved potency get to be synthesized. Also, if an analogue is found which defies the model, it suggests that some other factors are important and that provides a lead for further studies [39].
Several physicochemical parameters can be calculated in developing a QSAR model. However, the most common parameters used are hydrophobic, electronic and steric properties [40]. Due to the complexity that could emerge in calculating all these properties simultaneously and relating them to biological activity, each of the properties is varied one at a time while the rest is kept constant. In a simple case, several compounds with varying physicochemical properties (e.g. log P) are prepared and tested to investigate how these affect the biological activity (log 1/C). A graph of log 1/C vs log P is plotted and with the help of statistical tool (usually linear regression analysis by least square method), QSAR equations/models are developed. The regression or correlation coefficient (r) is a measure of how well the physicochemical parameter present in the equation explains the observed variance in biological activity. r values ≥ 0.9 are considered good fit while for regression coefficient quoted as r2, r2 ≥ 0.8 is taken as a good fit. Other statistical measures calculated to ensure goodness of fit include standard deviation, F-tests, etc.
2.8.1 Hydrophobicity
Drugs have to cross lipid-soluble regions such as cell membranes and fatty tissues, compete with metabolism process, which are fast for lipophilic drugs and with excretion process which are fast for water-soluble drugs before getting to their receptor [41]. Therefore, hydrophobic/lipophobic character of drugs play vital role to their biological effects, hence, it is necessary to predict this quantitatively. Hydrophobic property of a drug is determined experimentally by testing the drug’s relative distribution in an n-octanol/water mixture vis-à-vis its biological activity.
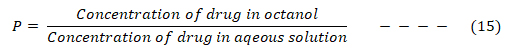
Where P is the relative distribution also known as partition coefficient
The biological activity is generally expressed as 1/C, where C is the concentration of drug required to achieve a defined level of biological activity. The reciprocal of the concentration (1/C) is used because more active drugs will achieve a defined biological activity at lower concentration.
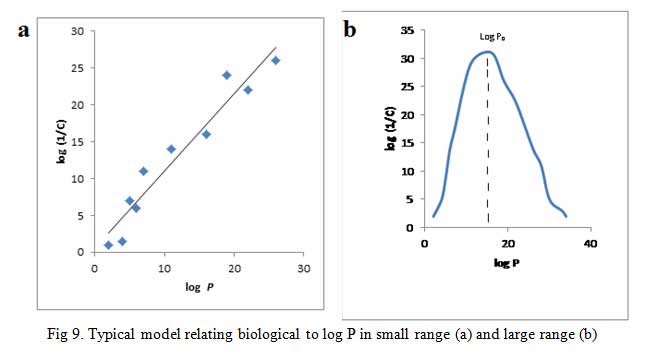
A plot of log (1/C) vs log P gives a straight line (Fig 9a) for cases with small range of log P, i.e. log P = 1-4, give equation 16:
![]()
Where K1 and K2 ![]()
![]() are proportionality constants.
are proportionality constants.
Click to view
Parabolic curve is obtained when biological activity is plotted against log P when P is large (Fig 9b) [42]. If the partition coefficient is the only factor influencing biological activity, the parabolic curve can be expressed by the equation (17):
![]()
Take for example QSAR equation derived when a set of large nonspecific, nonionic substances comprising of alcohols, ethers, and amides were tested for a narcotic effect on tadpoles.
![]()
The equation shows that fat solubility determines the accumulation of molecules in the nerve tissues of the tadpole which in turn influences their anesthetic potency. The r value indicates that the line resulting from the equation is a good fit. All the substances examined have their log P values on the upward slope of the parabolic curve. It would seem as though continuous increase in the log P will bring about an unending increase in biological effect. This is not so rather an optimum value of log P given in Fig 9b as log P0 is observed. In fact, any further increase in log P after log P0 results in declining biological effect.
Consider the anesthetic action of ethers whose mechanism of action do not involve drug-receptor interaction but solely on their ability to cross lipid membranes (cell membranes). The QSAR model/equation (19) for the ethers was generated as given below:
![]()
This equation can then be used to predict the anesthetic property of novel compounds. None the less, it should be noted that each QSAR model only applies to a series of compounds which have the same general structure. In the example, the model is derived solely for anesthetic ethers and therefore not applicable to other structural type of anesthetics.
The current progress in this area now makes it possible to calculate log P by computing the contributions that various substituents make to hydrophobicity. This helps to save resources and time because only substituents which make positive contribution to log P is synthesized.
2.8.2 Electronic Effect
The ionization and polarity of a drug molecule are influenced by the electronic state (electron withdrawing and donating property) of its various substituents. This in turn could affect drug’s transport to the receptor neighborhood and the drug-receptor interaction. For aromatic rings, the measure of electronic withdrawing and donating ability of a substituent is given by σ, known as Hammett substituent constant. The σx for a particular substituent (x) is defined by the equation:
![]()
Where KH is the equilibrium or dissociation constant; subscript H signifies that there are no substituents on the aromatic ring. Kx is dissociation constant of the analogue bearing the x substituent. Note that Kx can either be smaller than KH which happens when the substituent is electron donating group and vice versa. This leaves σx either as negative or positive values respectively.
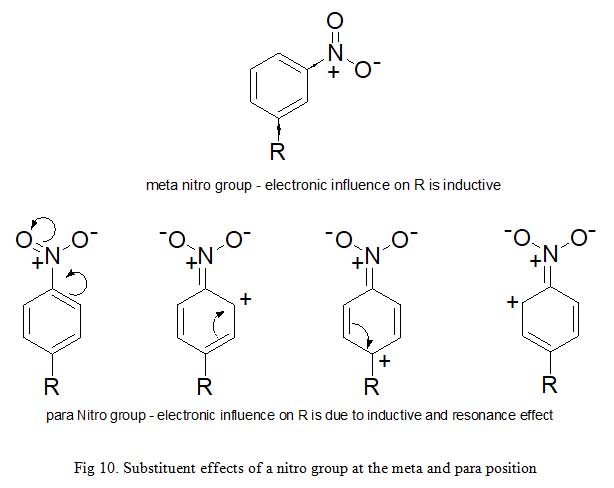
Due to the fact that σx takes resonance and inductive effects into account, its values depends on the position of the substituent on the parent aromatic compound. σp and σm symbolize substituents at para and meta positions respectively. For example; the electron withdrawing power of nitro group in meta-nitrobenzene is due to inductive effect only (σm = 0.71). But in para-nitrobenzene, both inductive and resonance influence participate (σp = 0.78). The same account for the discrepancies in Hammett substituent constant observed for hydroxyl group at meta (σm = 0.12) and p (σp = -0.37) positions. The inability of σx to measure for ortho substituents places a limitation to the electronic constants described so far, hence, substituents have an important steric and electronic effects. Secondly, only very few drugs are entirely influenced by electronic effect (Fig 10).
Click to view
2.8.3 Steric Factors
Many methods have been used to describe the influence of bulk, size and shape of a drug and these properties affect how it interacts with a binding site. Examples of such methods include: Taft’s steric factor (Es), molar refractivity and verloop steric parameter [43-44].
Taft’s steric factor (Es) value is calculated using the equation (21):
Es = log kx – log ko – – – – – (21)
Where kx and ko are the rate of hydrolysis of an aliphatic ester bearing the substituent x and the reference ester.
Taft’s steric factor only applies or is used to calculate the steric feature of the substituents which interact sterically with the tetrahedral transition state of the reaction and not by resonance or internal hydrogen bonding.
2.8.4 Hansch Equation
In reality, it is an uphill task to keep other parameters constant while calculating one physicochemical parameter as it is implied in the above equations. Therefore, there is need to develop method that can combine a number of different parameters and relate them to biological activity. The equation that does so is known as Hansch equation(22) [39]:
log 1/C = k1logP + k2σ + k3Es + k4 – – – – – (22)
This Hansch equation (linear) only applies when hydrophobicity value is in small range. For log P spread across a large range, the equation becomes parabolic as shown in equation (23):
log 1/C = – k1(logP)2 + k2logP + k3σ + k4Es + k5 – – – – – (23)
It has become necessary to consider the relative properties of different substituents by plotting a graph one physicochemical property against the other such as Craig plot in which the hydrophobicity substituent constant (π) is plotted against Hammett substituent constant (σ). Craig plot shows the relationship between the considered properties.
2.8.5 The Free-Wilson Approach
This method makes use of the overall effect of substituents to biological activities in constructing QSAR model. Unlike other approaches we have seen that calculate various physicochemical properties, this approach only requires experimental measurements of biological activity [45]. Biological activity of a parent structure is measured then compared with the activities of a range of substituted analogues. An equation (24) is then derived which relates biological activity to the presence, or otherwise, of particular substituents (X1-Xn).
Activity = k1X1 + k2X2 + k3X3 + … + knXn + Z – – – – – (24)
Where Xn is an indicator variable and has the value of 1 when the substituent “n” is present and the value of zero (0) when “n” is absent. kn values represent the contribution that each substituent makes to the activity. The average activity of the studied structures is given by the constant Z.
Free-Wilson approach is useful in quantifying specific molecular features which cannot be tabulated or the effect of unusual substituents that are not listed in the tables. In addition, the method can work in tandem with Hansch equations in developing an effective QSAR model as exemplified in developing QSAR equation for anti-allergic activity of a series of pyranenamines.
Three disadvantages have been identified in Free-Wilson method which are; this approach requires large number of analogues to be synthesized and tested in order to derive a meaningful equation. Also, the difficulty in rationalizing the results and explaining the reason a substituent’s position favours or disfavours activity. Another limitation is, the effects of different substituents may not be additive due to the occurrence of intramolecular interactions affecting activity.
The ability to predict a biological activity is valuable in any number of industries. Whilst some QSARs appear to be little more than academic studies, there are a large number of applications of these models within industry, academia and governmental (regulatory) agencies. A small number of potential uses are listed below:
- The rational identification of new leads with pharmacological, biocidal or pesticidal activity.
- The optimization of pharmacological, biocidal or pesticidal activity.
- The rational design of numerous other products such as surface-active agents, perfumes, dyes, and fine chemicals.
- The identification of hazardous compounds at early stages of product development or the screening of inventories of existing compounds.
- The designing out of toxicity and side-effects in new compounds.
- The prediction of toxicity to humans through deliberate, occasional and occupational exposure.
- The prediction of toxicity to environmental species.
- The selection of compounds with optimal pharmacokinetic properties, whether it be stability or availability in biological systems.
- The prediction of a variety of physico-chemical properties of molecules (whether they be pharmaceuticals, pesticides, personal products, fine chemicals, etc.).
- The prediction of the fate of molecules which are released into the environment.
- The rationalization and prediction of the combined effects of molecules, whether it be in mixtures or formulations.
The key feature of the role of in silico technologies in all of the listed areas of applications is that; predictions can be made from molecular structure alone.
2.9 Pharmacokinetic Profile of Drugs
It has become imperative to determine the absorption, distribution, metabolism and elimination properties, or better still ADMET, ADME/T or ADME/Tox (when considerations are given to toxicity issues) of potential drug molecules ab initio of drug design because many drugs fail to enter the market as a result of poor pharmacokinetic profile. In silico approaches are employed in the evaluation of these properties due to their cost and time effectiveness relative to experimental methods [46].
The well-known Lipinski’s Rule of Five was proposed to assess the oral bioavailability of a compound. It defines cutoffs for molecular mass, lipophilicity, number of hydrogen bond donors and acceptors which captures 90% of oral drugs and clinical candidates. Despite the fact that Rule of Five is often used as a filter, a number of drug compounds violate one or more criteria, therefore this should be kept as a guideline for bioavailability estimation. As mentioned in the Lipinski’s Rule of Five, the importance of lipophilicity and molecular size is widely accepted. Leeson and Springthorpe observed that high instability, as well as other ADMET liabilities including low solubility, high plasma protein binding (PPB), and high hERG (human ether-ago-go-related potassium channel protein) inhibition, can be caused by high lipophilicity. Low solubility and high PPB leads to low free plasma concentration of the compounds, which is critical to getting the desired pharmacology effect. HERG is a voltage-gated potassium ion channel, and high inhibition of this protein can cause QT prolongation which may result in fatal cardiac arrhythmia. Analysis of several simple physicochemical properties showed that drugs have significantly lower lipophilicity and molecular sizes than general bioactive compounds, compounds which have effects on living systems, and clinical candidates [47].
African trypanosomiasis is a parasitic disease that poses a significant health challenge to both humans and animals. Chemotherapy for the disease is poor. Except for fexinidazole, which is still in phase II/III clinical study in patients with late-stage of the disease, the only four drugs (suramin, melarsoprol, pentamidine and eflornithine) currently used to treat trypanosmiasis were developed about 30 years ago. Besides, each of these drugs has one or more of these challenges: expensive, highly toxic and require parenteral administration. Therefore, there is an urgent need for new and improved drugs against trypanosomal infection. Natural products from African rich vegetation with promising antitrypanosomal potencies have not resulted to drugs/drug leads because of high cost and time demand in drug development. Nowadays, these two limitations are greatly minimized by the use of computational techniques.
2.10 Successes of Computer-Aided-Drug-Design
SBDD has played vital role in drug design and development as shown in the following examples [48-49]:
- Inhibitors of Hsp90. Hsp90 is considered an essential therapeutic target for oncology because as a molecular chaperone, it modulates the activity of multiple oncogenic processes. 700,000 compounds from rCat were docked towards Hsp90 to identify potential hits (Hsp90 inhibitors). 900 nonredundant hits were identified from the virtual screening effort and 719 of the 900 compounds were purchased and tested in vivo which yielded 13 and 7 compounds with IC50< 100 µM and < 10 µM respectively. A study of hit-protein complex identified resorcinol-pyrazole series of compounds as lead and upon structural optimization of the lead arrived at compound AUY922 which then was investigated for myeloma, breast, lung and gastric cancers.
- Discovery of M1 Acetylcholine Receptor Agonists. Selective agonist of M1 mAChR is used to treat dementia such as Alzheimer’s disease and cognitive impairment associated with schizophrenia. Budzik et al. [48] virtually screened a collection of corporate compounds against this target to arrive at 1000 putative hits. Following in vitro assay, optimization for improving potency and selectivity for M1 mAChR, a series of novel 1-(N-substituted piperidin-4-yl) benzimidazolones were developed which have shown potency (as M1 mAChR agonist), ability to permeate the CNS and are drug-like.
3 Conclusion
The use of computers and molecular modeling strategies has become integral part of modern drug design and development techniques. This is majorly because of the time and cost effectiveness of the method. Also, the increasing agreement existing between computational and experimental results validates this research technique. However, continuous research is still ongoing to improve accuracy and speed of current software and algorithms used in computation.
4 Acknowledgments
The support by Dr. F. Ntie-Kang (Department of Chemistry, Chemical and Bioactivity Information Centre, Faculty of Science, University of Buea, Cameroon) and Dr. O.O. Olubiyi (Department of Pharmaceutical Chemistry, Obafemi Awolowo University, Ile Ife, Nigeria) is gratefully acknowledged.
5 References
- Balbes L, Mascarella S, Boyd D. A perspective of modern methods in computer-aided drug design. In: Lipkowtic K and Boyd DB, editors. Reviews Comput Chem. VCH Publishers; 1994, 5, 337-370.
- Shaikh SA, Jain T, Sandhu G, Latha N, Jayaram B. From drug target to leads – sketching a physicochemical pathway for lead molecule design in silico. Curr Pharm Des 2007; 13(34): 3454-3470.
- Shekhar C. In Silico Pharmacology: Computer-aided methods could transform drug development. Chem Biol 2008; 15: 413-414.
- Lengauer T. Algorithmic research problems in molecular bioinformatics. In IEEE Proceedings of the 2nd Isreali Symposium on the Theory of Computing and Systems, 1993; 177-192.
- Richards WG. Computer-aided drug design. Pure Appl Chern 1994; 66(8): 1589-1596.
- Hubbard RE. Can drugs be designed? Curr Opinion Biotech 1997; 8: 696-700.
- Topliss J (ed) Quantitative structure activity relationship of drugs. New York, Academic Press, 1983.
- Cramer RD, III, Redl G, Berkoff CE. Substructural analysis: A Novel Approach to the Problem of Drug Design. J Med Chem 1974; 17: 533-535.
- Ahindra Nag, Baishakhi Dey. Computer-Aided Drug Design and Delivery Systems. The McGraw-Hill Companies, Inc, 2011.
- Parsons J, Holmes JB, Rojas JM, Tsai J, Strauss CE. Practical conversion from torsion space to cartesian space for in silico protein synthesis. J Comput Chem 2005; 26: 1063-1068.
- Leach A. In Reviews in Computational Chemistry, Eds. Lipkowtz K and Boyd D. VCH Publishers, 1991; 2: 1-47.
- Zhen, Shu; Davies GJ. “Calculation of the Lennard-Jones n–m potential energy parameters for metals”. Physica Status Solidi (a), 1983; 78(2): 595–605.
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J Comp Chem 1983; 4: 187–217.
- Abraham DJ, Burger A. Burger’s medicinal chemistry and drug discovery, 6th edn., Wiley, Hoboken, N.J., 2003.
- Rao BG. Recent developments in the design of specific matrix metalloproteinase inhibitors aided by structural and computational studies. Curr Pharm Des 2005; 11(3): 295-322.
- Ntie-Kang F, Nwodo NJ, Ibezim A, Simoben CV, Karaman B, Ngwa VF, Sippl W, Adikwu MU, Mbaze ML. Molecular Modeling of Potential Anticancer Agents from African Medicinal Plants. J Chem Inf Model. 2014; 54: 2433−2450.
- Leonardo GF, Ricardo NS, Glaucius O, Adriano DA. Molecular Docking and Structure-Based Drug Design Strategies. Molecules, 2015; 20: 13384 – 13421.
- Huang N, Shoichet BK, Irwin JJ. Benchmarking sets for molecular docking. J Med Chem 2006; 49: 6789-6801.
- Glen RC, Allen SC. Ligand protein docking cancer research at the interface between biology and chemistry. Curr Med Chem 2003; 10: 767-782.
- Jones G, Willett P, Glen RC, Leach AR, Taylor A. Development and validation of a genetic algorithm for flexible docking. J Mol Biol 1997; 267: 727-748.
- Zsoldos Z, Reid D, Simon A, Sadjad SB, Johnson AP. eiHiTS: a new fast, exhaustive flexible ligand docking system. J Mol Graphics Model 2007; 26: 198-212.
- Leach AR, Shoichet BK, Peishoff CE. Prediction of protein ligand interactions. Docking and scoring: successes and gaps. J Med Chem 2006; 49: 5851-5855.
- Moitessier N, Englebienne P, Lee D, Lawandi J, Corbeil CR. Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go. Br J Pharmacol 2008; 153: S7-S26.
- Roughley S, Wright L, Brough P, Massey A, Hubbard RE. Hsp90 inhibitors and drug from fragment and virtual screening. Top Curr Chem 2012; 317: 61-82.
- Rinehart KL, Holt TG, Fregeau NL, Stroh JG, Keifer PA, Sun F, Li LH, and D. G. Martin DG, J Org Chem. 1990; 55: 4512-4515.
- Alonso H, Bliznyuk AA, Gready JE. Combining docking and molecular dynamic simulations in drug design. Med Res Rev 2006; 26: 531–568.
- Chen YC. Beware of docking! Trends Pharmacol Sci 2015; 36: 78–95.
- Salsbury FR Jr. Molecular dynamics simulations of protein dynamics and their relevance to drug discovery. Curr Opin Pharmacol. 2010; 10: 738–744.
- Durrant JD, McCammon JA. Molecular dynamics simulations and drug discovery. BMC Biol 2011; 9.
- Becker OM, Mackerell Jr AD, Roux B, Watanabe M. Computational biochemistry and biophysics. Marcel Dekker, Inc. 2001, 270 Madison Avenue, NY.
- Olubiyi OO, Frenzel D, Bartnik D, Gluck JM, Brener O, Nagel-Steger L, Funke SA, Willbold D, Strodel B. Amyloid aggregation inhibitory mechanism of arginine-rich D-peptides. Curr Med Chem. 2014; 21(2): 1448-57.
- Navia MA, Murcko MA. Use of structural information in drug design. Curr Opinion in Structural Biol 1992; 2: 202-216.
- Yang SY. Pharmacophore modeling and applications in drug discovery: challenges and recent advances. Drug Discov Today 2010; 15: 444-450.
- Wolber G, Langer T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their uses as virtual screening filters. J Chem Inf Model 2005; 45: 160-169
- Sutherland JJ, O’Brien LA, Weaver DF. A comparison of methods for modeling quantitative structure activity relationships. J Med Chem, 2004; 47: 5541-5554.
- Cramer RD III, Snader KM, Willis CR, Chakrin LW, Thomas J, Sutton BM. Application of quantitative structure activity relationships in the development of the antiallergenic pyranenamines. J Med Chem. 1979; 22: 714-725.
- Kubini H, Folkers G, Martin YC (eds) 3D QSAR in Drug Design. Kluwer/Escom, Dordrecht, 1998.
- Hansch C, Leo A. Exploring QSAR. Amer Chem Soc, Washington, DC, 1995.
- Leach AR. Molecular modeling: principles and applications. 2nd edn. Pearson Education, London, 2001.
- Ibezim A, Onyia K, Ntie-Kang F, Nwodo NJ. Drug-like properties of potential anti-cancer compounds from Cameroonian flora: A virtual study. J Applied Pharm Sci 2015; 5(06): 133-137.
- Kellogg GE, Abraham DJ. Hydrophobicity: Is log P more than the sum of its parts? Eur J Med Chem 2000; 35: 651-661.
- Taft RW. Separation of polar, steric and resonance effects in reactivity. In: Newman MS (ed) Steric Effects in Organic Chemistry. John Wiley and Sons, New York, 1956.
- Varloop A, Hoogenstraaten W, Tipker J. Development and application of new steric substituent parameters in drug design. Med Chem. 1976; 11: 165-207.
- Van de Waterbeemd H, Testa B, Folkers G (eds). Computer-assisted Lead Finding and Optimization. Wiley-VCH, New York, 1997.
- Onoabedje EA, Ibezim A, Okafor SN, Onoabedje US, Okoro UC. Oxazin-5-Ones as a Novel Class of Penicillin Binding Protein Inhibitors: Design, Synthesis and Structure Activity Relationship. PLoS ONE, 2016; 11(10): e0163467.
- Ibezim A, Mbah CJ, Ntie-Kang F, Adikwu MU, Nwodo NJ. Virtual screening of Trypanosoma Inhibitors from Nigeria. J Cheminformatics 2016; 8(Suppl 1):P12
- Dictionary of natural products on CD-rom. Chapman and Hall/CRC Press, London, 2005.
- Budzik B, Garzya V, Walker G, Woolley-Roberts M, Pardoe J, Lucas A, Techan B, Rivero RA, Langmead CJ et al. Novel N-substituted benzimidazolones as potent, selective, CNS-penetrant and orally active M(1) mAChR agonists. Acs Med Chem Lett 2010; 1: 244-248.
- Schneider G, Fechner U. Computer-based de novo design of drug-like molecules. Nat Rev Drug Discov 2005; 4: 649-663.